TagCleaner. For cleaner sequences.
Manual
Web versionNecessary resources
Upload data
Standalone version
Necessary resources
Available options
Examples
Notes
Tag sequence trimming
Allowed mismatches
Trimming range
Continuous trimming
Non-matching reads
Concatenated reads
Notes

Preprocessing 454 data
01/16/2011
PDF, 92 KB

Preprocessing FASTA-only data
01/16/2011
PDF, 76 KB

Raw data processing
01/16/2011
PDF, 112 KB

Quality control
01/16/2011
PDF, 520 KB

Data preprocessing
09/01/2011
PDF, 827 KB
Introduction
Sequencing metagenomes that were pre-amplified with primer-based methods requires the removal of the additional tag sequences that can contain deletions or insertions due to sequencing limitations. The tag (e.g. WTA primer) sequence may be unavailable or incorrectly reported in public databases. Because of the potential for downstream inaccuracies introduced by unwanted sequence contaminations, it is important to use reliable tools for pre-processing sequence data.
TagCleaner is distributed under the GNU Public License (GPL). All its source codes are freely available to both academic and commercial users. The latest version can be downloaded at the SourceForge download page.
Web version
TOP OF PAGEThe interactive web interface of TagCleaner can be used to automatically detect and efficiently remove tag sequences from genomic and metagenomic datasets.
Necessary resources
Hardware
- Computer connected to the Internet
Software
- Up-to-date Web browser (Firefox, Safari, Chrome, Internet Explorer, ...)
Files
- FASTA file with sequence data
- FASTQ file (as alternative format to trim sequence and quality data)
Upload data to the TagCleaner web version
To upload a new dataset in FASTA or FASTQ format to TagCleaner, follow these steps:
1. Go to http://tagcleaner.sourceforge.net
2. Click on "Use TagCleaner" in the top menu on the right (the latest TagCleaner web version should load)
3. Select your FASTA or FASTQ file
4. Select trim mode (trim tag sequences from both ends, 5'-end only, or 3'-end only)
5. Specify tag sequence for 5'-end and/or 3'-end (if available)
6. Click "Submit"
Notes
If the tag sequences are not available or are unknown, leave the fields free and TagCleaner will try to find the tag sequence. The predicted tag sequence is provided to the user and can be modified before any further steps are performed (see below).
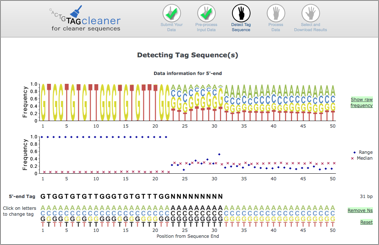
TagCleaner does not require the setting of filter parameters (such as maximum number of mismatches) before the data is processed. Instead, the filter parameters are set after the data is processed, which allows the user to choose parameters appropriate for their dataset and does not require them to submit and process the same data with modified parameters for several times.
Tag sequence trimming
TOP OF PAGETag sequence trimming should be performed before quality trimming and sequence dereplication. The trimming of low-quality bases at the ends might truncate the tag sequence and reduce the ability to recognize the remainder of the tag sequence. In those cases, large parts of the tag sequences might still remain for further analysis and data processing steps. Dereplication before trimming may miss duplicated sequences due to variations in the tag sequences that will be trimmed off later and would therefore require an additional dereplication step after the trimming.
The algorithm implemented in TagCleaner for the automatic detection of tag sequences assumes the randomness of a typical metagenome. Datasets that do not contain random sequences from organisms in an environment, but rather contain, for example, 16S data may cause incorrect detection of the tag sequences. However, the tag sequences will most likely be over-predicted and can be redefined by the user prior to data processing.
Maximum allowed mismatches
The independent definition of maximum allowed mismatches for the 5'- and 3'-end of the reads accounts for the differences in tag sequences due to the limitations of the sequencing method used to generate the datasets. The 3'-end will in most cases show a lower number of matching tag sequences with low number of mismatches due to incomplete or missing tags at the ends of incompletely sequenced fragments.
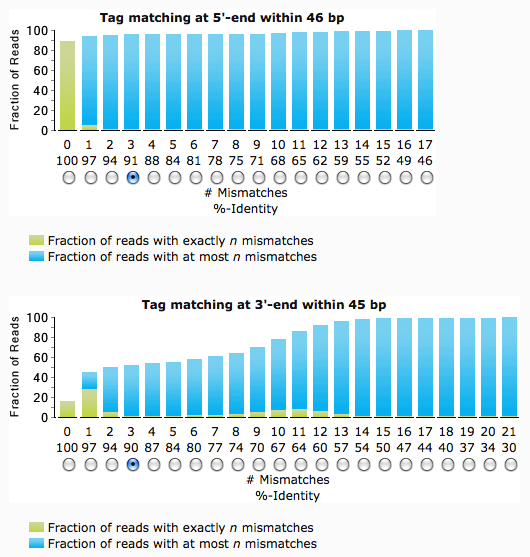
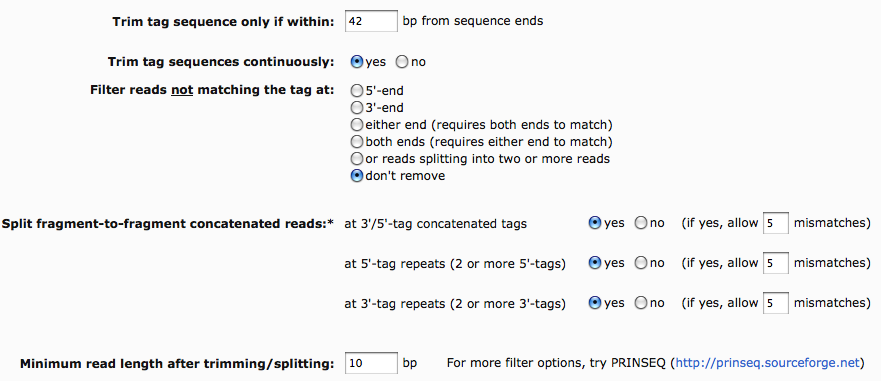
Trim tag sequence only if within
The sequence of the tag could occur not only at the sequence end, but also at any other position of the sequence. To assure that only tags are trimmed, the tag sequences can be defined to occur only at the ends allowing a certain number of variable bases. The default value for tag sequence of at least 10 bp is 1.5 times the tag sequence length.
Trim tag sequence continuously
The continuous trimming of tag sequences from the ends allows filtering of sequences mainly consisting of concatenated tag sequences.
Remove reads not matching tag sequence
This filter can be applied to remove sequences that do not contain any tag sequence (with the defined number of maximum mismatches). This feature was originally designed to separate MID tagged sequences before the MID tags were made publicly available. The feature can also be used to further investigate sequences without a matching tag sequence.
Split fragment-to-fragment concatenated reads
TagCleaner provides an additional feature of detecting and splitting fragment-to-fragment concatenations. This important pre-processing step removes tag contaminations inside the sequences, which may allow, for example, more accurate assemblies. The concatenated fragments may additionally present a source of error for annotation and taxonomic assignments, since fragments from different organisms may not be assigned correctly when concatenated.
Notes
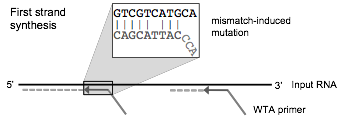
TagCleaner is able to detect the quasi-random 3'-end of whole transcriptome amplification (WTA) primers. The user has the option whether or not to trim this part of the tag sequence by simply adding or removing the letter N from the end of the tag sequence. However, users are advised to trim the complete tag sequence. It is important to trim the random parts in order to account for mismatch-induced mutations that often happen when primers anneal to similar (but not identical) sequences with high enough affinity for binding (see below). Therefore, one cannot be certain that this part of the tag sequence represents the actual sequence of the sample.
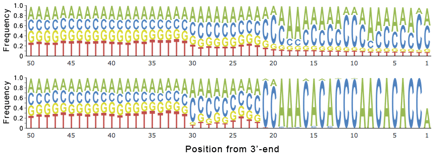
The tag sequence prediction uses filtered base frequencies instead of raw base frequencies. This allows a more accurate prediction as it accounts for incomplete and shifted tag sequences. The following example shows the raw base frequencies (top) and the filtered base frequencies (bottom) for the 3'-ends.
Standalone version
TOP OF PAGEThe standalone version does not provide graphical outputs, but all the functionality you might be used to from the web version. Starting with version 0.9, the web version is using the standalone version for all the calculations in the backend. The readme file contains information on the usage of the standalone version.
Necessary resources
Hardware
- Computer with a Linux/Unix or Mac OS X operating system
Software
- TagCleaner standalone (available at http://deconseq.sourceforge.net)
- Perl 5 (or higher)
Files
- FASTA file with sequence data (and optional QUAL file to trim sequences and their quality scores)
- FASTQ file (as alternative format)
Available options
|
Option/flag |
Description |
Default |
Range |
-help or -h |
Print the help message; ignore other arguments |
||
-man |
Print the full documentation; ignore other arguments |
||
-version |
Print program version; ignore other arguments |
||
-verbose |
Prints status and info messages during processing |
||
| Input options | |||
-fastq |
Input file in FASTQ format that contains the sequence and quality data. |
|
STRING |
-fasta |
Input file in FASTA format that contains the sequence data. |
|
STRING |
-qual |
Input file in QUAL format that contains the quality data. |
|
|
-matrix |
Use union, subset or exact matrix instead of default matrix when matching tag characters to the sequence characters. The default matrix requires that the ambiguous characters of the sequence represent bases that are a subset of the ambigous characters of the tag sequence (e.g. R = AG in the sequence is a subset of D = AGT and is marked with an X in the matrix). The subset matrix requires that either the tag character or the sequence character is a subset of the other. The union matrix requires that at least one base is common in tag and sequence (e.g. R = AG and M = AC have base A in common and are marked with an X in the matrix). The exact matrix allows to trim ambiguous bases without considering the ambiguity code (e.g. N in tag only trims N, not ACGT). Optional options are either "subset", "union" or "exact". |
Default matrix |
[subset, union] |
| Output options | |||
-out |
By default, the output files are created in the same directory as the input file containing the sequence data with an additional "_tagcleaner_XXXX" in their name (where XXXX is replaced by random characters to prevent overwriting previous files). To change the output filename and location, specify the filename using this option. |
|
STRING |
-out_format |
To change the output format, use one of the following options. If not defined, the output format will be the same as the input format. |
|
[1,2,3] |
-line_width |
Sequence characters per line. Use 0 if you want each sequence in a single line. Use 80 for line breaks every 80 characters. Note that this option only applies to FASTA output files, since FASTQ files store sequences without additional line breaks. |
60 |
INT |
-stats |
Prints the number of tag sequences matching for different numbers of mismatches. In combination with -split, the number of sequences with fragment-to-fragment concatenations is printed as well. Cannot be used in combination with -predict and will not perform any trimming. The output values are separated by tabs with the header line: "#Param Mismatches_or_Splits Number_of_Sequences Percentage Percentage_Sum". Cannot be used in combination with -predict and require -tag5 or -tag3. |
|
|
-predict |
Use this option to have TagCleaner predict the tag sequences. It will attempt to predict the tag at either or both sites, if possible. The algorithm implemented for the tag prediction assumes the randomness of a typical metagenome. Datasets that do not contain random sequences from organisms in an environment, but rather contain, for example, 16S data may cause incorrect detection of the tag sequences. However, the tag sequences will most likely be over-predicted and can be redefined by the user prior to data processing. The tag sequence prediction uses filtered base frequencies instead of raw base frequencies. This allows a more accurate prediction as it accounts for incomplete and shifted tag sequences. The output values are separated by tabs with the header line: "#Param Tag_Sequence Tag_Length Percent_Explained". If no tags are reported, then no tags could be identified in the data set. Cannot be used in combination with -tag3 or -tag5 or -stats. When using this option, no trimming will be performed. |
|
|
-log |
Log file to keep track of parameters, errors, etc. The log file name is optional. If no file name is given, the log file name will be "inputname.log". If the log file already exists, new content will be added to the file. |
|
FILE |
-info |
This option will provide the trimming and splitting information in the header line after the sequence identifier. The following information is given and separated by a single space: initial length, length after trimming, 5'-end trimming position, 3'-end trimming position, number of mismatches at 5'-end, number of mismatches at 3'-end and number of sequences after splitting. In case of a splitting event, the number of mismatches at the 5'- and 3'-end will be the number of mismatches of the concatenated tags. |
|
|
-nomatch |
This option allows to filter sequences that do not match the tag sequence at the ends or do not contain inner tags within the maximum number of allowed mismatches. The following options allow to filter reads not matching the tag at: |
|
[1,2,3,4,5] |
-minlen |
Minimum read length of trimming and/or splitting |
|
INT |
-filtered |
Output the sequences that would be filtered out instead of the sequences passing the filters. This includes sequences that e.g. are tag sequence repeats or do not fulfill the -nomatch option. |
|
|
| Trim/Split options | |||
-tag5 |
Tag sequence at 5'-end. Use option -predict if unknown. |
|
STRING |
-tag3 |
Tag sequence at 3'-end. Use option -predict if unknown. |
|
STRING |
-mm5 |
Maximum number of allowed mismatches at the 5'-end. |
0 |
INT |
-mm3 |
Maximum number of allowed mismatches at the 3'-end. The independent definition for the 5'- and 3'-end of the reads accounts for the differences in tag sequences due to the limitations of the sequencing method used to generate the datasets. The 3'-end will in most cases show a lower number of matching tag sequences with low number of mismatches due to incomplete or missing tags at the ends of incompletely sequenced fragments. |
0 |
INT |
-trim_within |
The sequence of the tag could occur not only at the sequence end, but also at any other position of the sequence. To assure that only tags are trimmed, the tag sequences can be defined to occur only at the ends allowing a certain number of variable bases. The default value for -trim_within for a tag sequence of at least 10 bp is 1.5 times the tag sequence length. |
1.5x tag |
INT |
-cont |
Trim tag sequences continuously. This is helpful if you have sequence with tag sequence repeats or sequence that are concatenated tag sequences. Note that a high number of allowed mismatches and continuous trimming can cause over-trimming. Use more than 20% mismatches with continuous trimming only with caution. |
|
|
-split |
This feature removes tag contaminations inside the sequences and splits fragment-to-fragment concatenations into separate sequences. The optional integer value specifies the maximum number of allowed mismatches for the internal (concatenated) tag sequence(s). This feature should be used with caution for inputs with only a 5' or 3' tag sequence (likely splits too many false positive that naturally occur for single tags compared to much longer concatenated 5' and 3' tags). The number of mismatches is used as maximum value for -stats. This option will cause a decrease in speed. |
0 |
INT |
-split5r |
This feature is similar to -split, but instead of search for 3'-5'-tag repeats, it will search for 5'-tag repeats (2 or more). This option only applies if both -tag3 and -tag5 are specified. To split at a single 5'-tag, run the program without -tag3 and with -split. |
0 |
INT |
-split3r |
This feature is similar to -split, but instead of search for 3'-5'-tag repeats, it will search for 3'-3'-tag repeats (2 or more). This option only applies if both -tag3 and -tag5 are specified. To split at a single 3'-tag, run the program without -tag5 and with -split. |
0 |
INT |
-splitall |
This features is for convenience only and applies the integer value to all split options (-split, -split3r and -split5r). This option only applies if both -tag3 and -tag5 are specified and will overwrite all other split options with the given integer value. |
0 |
INT |
Examples
The following examples apply to version 0.9 and higher and assume a 64 bit architecture. If you do not use a 64 bit system, ignore the -64 option.
Verbose mode (-verbose)
The verbose mode shows you what the program is doing while it is running and also the progress of tasks that require more computation:
$ perl tagcleaner.pl -verbose -64 -fastq SRR004254.fastq -predict
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
status: 34 %Predicting the tag sequence (-predict)
To predict the tag sequences, you can simply use the -predict option:
$ perl tagcleaner.pl -verbose -64 -fastq SRR004254.fastq -predict
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
#Param Tag_Sequence Tag_Length Percentage_Explained
tag3 NNNNNNNNNNCCAAACACACCCAACACA 28 49.52
tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN 28 71.56Using the non-verbose mode:
$ perl tagcleaner.pl -64 -fastq SRR004254.fastq -predict
#Param Tag_Sequence Tag_Length Percentage_Explained
tag3 NNNNNNNNNNCCAAACACACCCAACACA 28 49.52
tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN 28 71.56Note that the comment lines in the outputs will start with the pound sign (#) that can be eaily parsed by other programs. All values in a row are separated by tabs.
Using -predict will not trim any input sequences and can only be used to predict the possible tag sequence(s).
Checking for tag sequences (-predict)
The percentage values given in the column called "Percentage_Explained" (see example above) can be used to estimate the probability of a tag being real. If the percentage is high, there is likely a tag. If the percentage is low (5-10%), then there is likely no tag.
Using the stats option (-stats)
This options can be used to get the number of sequences that match the tag sequence(s) with different numbers of allowed mismatches.
$ perl tagcleaner.pl -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN -stats
#Param Number_of_Mismatches_or_Splits Number_of_Sequences Percentage Percentage_Sum
tag3 0 7167 46.85 46.85
tag3 1 316 2.07 48.91
tag3 2 696 4.55 53.46
tag3 3 348 2.27 55.74
tag3 4 556 3.63 59.37
tag3 5 227 1.48 60.86
tag3 6 255 1.67 62.52
tag3 7 431 2.82 65.34
tag3 8 1031 6.74 72.08
tag3 9 1595 10.43 82.51
tag3 10 1538 10.05 92.56
tag3 11 622 4.07 96.63
tag3 12 227 1.48 98.11
tag3 13 99 0.65 98.76
tag3 14 25 0.16 98.92
tag3 15 20 0.13 99.05
tag3 16 34 0.22 99.27
tag3 17 34 0.22 99.50
tag3 18 77 0.50 100.00
tag5 0 10752 70.28 70.28
tag5 1 533 3.48 73.77
tag5 2 462 3.02 76.79
tag5 3 492 3.22 80.00
tag5 4 258 1.69 81.69
tag5 5 103 0.67 82.36
tag5 6 104 0.68 83.04
tag5 7 231 1.51 84.55
tag5 8 371 2.43 86.98
tag5 9 628 4.11 91.08
tag5 10 775 5.07 96.15
tag5 11 325 2.12 98.27
tag5 12 180 1.18 99.45
tag5 13 25 0.16 99.61
tag5 14 8 0.05 99.67
tag5 15 16 0.10 99.77
tag5 16 5 0.03 99.80
tag5 17 8 0.05 99.86
tag5 18 22 0.14 100.00Note that the tag is shorter than 32 bp and therefore we do not need the -64 option, which is only required for longer tag sequences using more than the default 32 bit. The printed table tells us how many sequences match the tag3 or tag5 sequence with how many mismatches. The slash (/) allows to continue commands over multiple lines and is used here to keep the lines short enough for easy display.
Trimming tags (-tag5, -tag3, -mm5 and -mm3)
When you perform the tag trimming, the -log option will be useful to keep track of the processing of your data.
$ perl tagcleaner.pl -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN \
-mm5 3 -mm3 3 -verbose -log
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
Input/Output stats:
Input sequences: 15,298
Input bases: 3,122,243
Input mean length: 204.09
Output sequences: 15,298
Output bases: 2,538,321
Output mean length: 165.93The verbose mode will provide some basic statistics of the input and output data. Since we did not provide a filename for the output file and log file, it will be automatically generated. The output file in this example is "SRR004254_tagcleaner_t5gF.fastq" and the log file "SRR004254.fastq.log". The number of input and output sequences is identical, as we did not filter the trimmed sequences by matching tags or sequence length. The log file contains all the processing information and comes in handy if you perform multiple trimming or preprocessing steps. The log file for this example contains the following content:
[tagcleaner-standalone-0.9] [02/09/2011 15:55:00] Executing TagCleaner with command: "perl tagcleaner.pl -fastq SRR004254.fastq -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN -tag3 NNNNNNNNNNCCAAACACACCCAACACA -mm5 3 -mm3 3 -verbose -log"
[tagcleaner-standalone-0.9] [02/09/2011 15:55:00] Write results to file: "SRR004254_tagcleaner_Tnqj.fastq"
[tagcleaner-standalone-0.9] [02/09/2011 15:55:00] Parse and process input data: "SRR004254.fastq"
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Finished processing input data: "SRR004254.fastq"
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Input sequences: 15,298
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Input bases: 3,122,243
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Input mean length: 204.09
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Output sequences: 15,298
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Output bases: 2,538,321
[tagcleaner-standalone-0.9] [02/09/2011 15:55:05] Output mean length: 165.93Splitting fragment-to-fragment concatenations (-split)
The splitting option can be used to split fragment-to-fragment concatenations at 3'-5' concatenated tag sequences:
$ perl tagcleaner.pl -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN \
-mm5 3 -mm3 3 -split 5 -verbose
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
Input/Output stats:
Input sequences: 15,298
Input bases: 3,122,243
Input mean length: 204.09
Output sequences: 15,695
Output bases: 2,515,773
Output mean length: 160.29Note the difference in number of input and output sequences due to the splitting of concatenated fragments into separate sequences allowing 5 mismatches for the concatenated tags.
Splitting more complex concatenations (-split5r and -split3r)
Real example sequence with 3'-5', 5'-5' and 5'-5' concatenated tags:
ACGGGCTTAGTGATCCGGTGGTTCCGCATGGAAGGGCCATCGCTCAACGGATAAAAGCTACCCCGGGGATAACAGGCTTA
TCTCCCCCAAACCCCAAACACACCCAACACAGTGTTGGGTGTGTTTGGTGTGTGGTGGTTGTGTTGGGTGTGTTTGGTTT
TGGTTGGGTGTGTTGGGTGTGTTTGGGGTTGGTGGGCTGTGTTGGGTGTGTTTGGTGGTGTGTGGGTTGTGTTGGGTGTG
TTTGGGTATGCGTGGTAGGAGAGCGTTCTAACAGCGTTGAAGTCAGACCGGAAGGACT
To split fragments at more complex tag sequence concatenations such as in this worse case scenario "sequence1-tag3-tag5-tag5-tag5-sequence2-tag5-tag5-tag5-sequence3-tag3-tag3-sequence4", you would need to use additional options for splitting at tag sequence repeats:
$ perl tagcleaner.pl -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN \
-mm5 3 -mm3 3 -split 5 -split5r 5 -split3r 5 -verbose
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
Input/Output stats:
Input sequences: 15,298
Input bases: 3,122,243
Input mean length: 204.09
Output sequences: 16,479
Output bases: 2,454,259
Output mean length: 148.93Note the even higher number of output sequences suggesting that the data set contained fragment-to-fragment concatenated sequences that could be identified and split at tag sequence repeats.
Continuous trimming (-cont)
This option is useful to trim tag sequence repeats at the ends, as shown in the example below:TGTTGGGTGTGTTTGGTGTGTGGTGGTTGTGTTGGGTGTGTTTGGTTTTGGTTGGTGTGTTGGGTGTGTTTGGGGTTGGT
GGGCTAGGAGAGCGTTCTAACAGCGTTGAAGTCAGACCGGAAGGACTACGGGCTTAGTGATCCGGTGGTTCCGCATGGAA
GGGCCATCGCTCAACGGATAAAAGCTACCCCGGGGATAACAGGCTTATCTCCCCCAAACCCCAAACACACCCAACACA
$ perl tagcleaner.pl -verbose -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN -mm5 3 -mm3 3 -cont
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
Input/Output stats:
Input sequences: 15,298
Input bases: 3,122,243
Input mean length: 204.09
Output sequences: 15,280
Output bases: 2,386,486
Output mean length: 156.18Note the lower number of sequence that suggest that the data set contained sequences that were pure tag sequence repeats.
Trim tag sequences further from the ends (-trim_within)
This option is useful to trim tags that are not occoring right at the ends, as shown in the following example:AGCGTAGTCGATGGGAAACAGGTTAATATTCCTGTACTTCTGGTTACTGCGATGGAGGGACGGAGAAGGCTAGGCCAGCT
TGGCATTGGTTGTCCAAGTTTAAGGTGGTAGGCTGAAATCTTAGGTAAATCCGGGGTTTCAAGGCCGAGAGCTGATGACG
AGTCGTCTTTTAGATGACGAAGTGGTTGATAGACCATGCTTCCAAGAAAAGCTTCTAAGCTTCAGGTAACCACGAAACCA
AACACACCCAACACACCAACAAAAAAAAAAAAAAAAAAAAAAAAAAACAAAAACAA
The 3'-tag can be found within 70 bp from the end and therefore, can be trimmed by using the -trim_within option with parameter 70.
$ perl tagcleaner.pl -verbose -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN -mm5 3 -mm3 3 \
-trim_within 70Filtering short reads (-minlen)
This option is useful to filter reads that are very short after trimming tag sequences and/or splitting.
$ perl tagcleaner.pl -verbose -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN -mm5 3 -mm3 3 \
-minlen 10Suggested options for trimming and splitting
This are the options that would be used in a good use case (the parameters are up to the specific use and should not be blindly copied without thinking about what they do):
$ perl tagcleaner.pl -verbose -fastq SRR004254.fastq \
-tag3 NNNNNNNNNNCCAAACACACCCAACACA -tag5 TGTGTTGGGTGTGTTTGGNNNNNNNNNN -mm5 3 -mm3 3 \
-splitall 5 -minlen 10 -cont -log SRR004254_preprocessing.log -out SRR004254_trimmed
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
Input/Output stats:
Input sequences: 15,298
Input bases: 3,122,243
Input mean length: 204.09
Output sequences: 15,662
Output bases: 2,354,964
Output mean length: 150.36This will provide you the basic statistics of the data processing using the verbose mode and writes all the processing steps to the file "SRR004254_preprocessing.log" by using the -log option and the trimmed and split data to file "SRR004254_trimmed.fastq" by using the -out option:
$ ls
SRR004254.fastq
SRR004254_preprocessing.log
SRR004254_trimmed.fastq
tagcleaner.plNotes
The following is a collection of random notes that might help you when using TagCleaner to process your data.
How to handle cases where tags are not completely sequenced
Depending on the length of your tag sequence, you have different options on how to deal with incomplete tag sequences during removal. If your tag sequences are long enough (e.g. 15+bp), then you could perform the search for end tags with the last 7-10 bases and specify that they have to be found only at the end (e.g. first/last 15 bases). If you have short primer sequences, then I can only suggest to slightly increase the number of mismatches.
An example command for a tag with the sequence ACCGTGACGTCTGAGACT (18 bases) could be:
perl tagcleaner.pl -log file.log -verbose -fastq file.fastq -tag3 ACCGTGAC -mm3 1 -trim_within 18 -out file_tagcleanedFor this approach, a low number of allowed mismatches should be defined to reduce the number of false positive trimmed tag sequences. Also, it might be useful to run TagCleaner first with the full tag sequence and a higher number of allowed mismatches and then again with the partial sequence and low number of allowed mismatches.
How to trim ambiguous bases without considering the ambiguity code
If you want to trim NNA from the 3'-end, but do not want to trim AAA or ATA, then you should use the matrix exact.
perl tagcleaner.pl -log file.log -verbose -fasta file.fasta -tag3 NNA -mm3 0 -trim_within 3 -matrix exact -out file_tagcleanedAllowing no mismatches and only checking at the last three positions will only trim NNA from the sequences and nothing else. If you also want to trim sequences with NNAA (and NNAC, NNAG, NNAT and NNAN), then you only have to modify the -trim_within parameter.
perl tagcleaner.pl -log file.log -verbose -fasta file.fasta -tag3 NNA -mm3 0 -trim_within 4 -matrix exact -out file_tagcleaned