TagCleaner. For cleaner sequences.
Detect and trim your tag sequences
from your sequence data.
The TagCleaner tool can be used to automatically detect and efficiently remove tag sequences (e.g. WTA tags) from genomic and metagenomic datasets. It is easily configurable and provides a user-friendly interface.
Input interface
The user can either input a data ID to access already processed data or input a new sequence file and the tag sequences, if available.
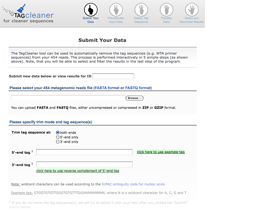
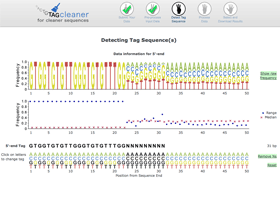
Interactive tag prediction
The estimated tag sequence is shown below the nucleotide frequency plot and frequency range and median plot. The user can change the tag sequence using the functionality of the graphical interface.
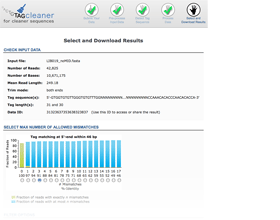
Output interface
The output interface provides input information, a graphical representation of the number of mismatches, and filter and download options.
News and Updates
Here you will find release notes and news related to TagCleaner.
10 / 2013
Release of version 0.16: Support tag prediction in mixed-case sequences.
Release of version 0.15: Fix warnings during tag prediction.
08 / 2013
Release of version 0.14: Fixed issue with line width parameter not being set. Fix warnings with defined statements.
06 / 2013
Release of version 0.13: Fixed issue that generated warnings when the trimmed sequence is zero-length. Modified filename check to prevent endless loop. Other minor fixes.
10 / 2011
Release of version 0.12: Added matrix "exact" to allow trimming of ambiguous bases without considering the ambiguity code.
09 / 2011
Barcode splitter: Added a Perl script to separate FASTA/FASTQ files by barcodes allowing errors. The file and readme is avilable under Downloads -> misc.
04 / 2011
Release of version 0.11: Added support for ambiguous characters in the input sequences (thanks to Dave Messina from Stockholm for the suggestion). Character matching can be set based on three predefined matrices (new option -matrix). Non-IUPAC characters for nucleic acids are mismatched by default.
03 / 2011
Release of version 0.10: Fixed issue in tag detection for unknown shifts. Added File::Path for legacy support.
02 / 2011
Release of version 0.9: First release of standalone version. Web version now uses standalone version in backend. Improved tag detection. New features for the splitting of concatenated fragments and tag repeats. Filter options are now limited as filtering should be performed using PRINSEQ tool after trimming. Web version now allows FASTQ input and output and the processing of larger input files.
09 / 2010
Release web-0.7: Fixed issue in tag detection when no shifts can be found and for parsing FASTQ files with no information in '+' header line.
06 / 2010
Release web-0.6: Added new function to tag sequence detection interface: "Use as 3'-end". Added Javascript function to check for tag sequence length to restrict tag sequences to max 64 bp.
05 / 2010
Release web-0.5: Fixed "substr outside of string" during detection of tag sequence repeats.
Release web-0.4: Fixed cross browser issues. Added new functions to tag sequence detection ("Reset to prediction" and "Clear all").